[拼音]:peiweichang lilun
[外文]:ligand field theory
说明和解释配位化合物的结构和性能的理论。在有些配合物中,中心离子(通常也称中心原子)周围被按照一定对称性分布的配位体所包围而形成一个结构单元。配位场就是配位体对中心离子(这里大多是指过渡金属络合物)作用的静电势场。由于配位体有各种对称性排布,遂有各种类型的配位场,如四面体配位化合物形成的四面体场,八面体配位化合物形成的八面体场等。
有三种理论用于说明和解释配合物的结构和性能:价键理论、晶体场理论和分子轨道理论。由L.C.鲍林提出的价键理论,在说明配合物(或配离子)的几何构型和磁学性质是很有效的(表1),但对含有较多的d电子的过渡金属配合物的稳定存在和配合物的电子光谱却不能给予满意的解释。因此,目前价键理论已基本不用了。而处理离子型配合物的晶体场理论和处理共价型配合物的分子轨道理论的结合成为配位场理论,在20世纪50年代以来得到迅速发展,用于讨论过渡金属配合物的物理化学性质。晶体场理论和分子轨道理论则是配位场理论的两种极限情况。
 晶体场理论
晶体场理论
由H.A.贝特和J.H.范扶累克提出的一种点电荷模型,认为配位体与中心离子的作用类似于离子晶体中正、负离子的静电吸引力,而不考虑中心离子的轨道与配位体轨道的重叠。
在无配位场存在下的中心离子是自由的,其电子云分布是球形对称的,五个d原子轨道处于同一个能级,这叫简并态。当配合物形成,即存在配位场的作用下,这些d轨道能级就要发生分裂(即部分消除简并),一部分能级处于比原能级高的位置,另一部分能级则处于比原能级低的位置,这称为能级分裂。例如CoF咶配离子,在6个氟离子形成的八面体场作用下,过渡金属离子的d轨道能级分裂为两组(图1 ),

能级较高的一组有两个d轨道(d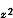 、d
、d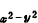 ),这组双重简并的d轨道用符号eg表示;另一组能级较低,有三个d轨道(dxy、dxz、dyz),用t2g标记。这两组轨道的能级差用墹表示,称为场分裂值。图1表明八面体场和四面体场的d轨道能级分裂情况。不同对称性的场分裂值墹是不同的,它的大小与中心离子和配位体的性质有关,按照理论计算和总结过渡金属络合物的电子光谱数据,可得出以下经验规律:
),这组双重简并的d轨道用符号eg表示;另一组能级较低,有三个d轨道(dxy、dxz、dyz),用t2g标记。这两组轨道的能级差用墹表示,称为场分裂值。图1表明八面体场和四面体场的d轨道能级分裂情况。不同对称性的场分裂值墹是不同的,它的大小与中心离子和配位体的性质有关,按照理论计算和总结过渡金属络合物的电子光谱数据,可得出以下经验规律:
(1)当中心离子固定时,墹 随下列配位体依次增加:
I-<Br-<SCN-<Cl-<F-<H2O<NCS-
<NH3<乙二胺<邻联吡啶<CN-
(2)当配位体固定时,墹 随下列中心离子依次增加:
Mn2+<Co2+<Ni2+<V2+<Fe3+<Cr3+<Co3+<Ru3+
<Mo3+<Rh3+<Pd4+<Ir3+<Re4+<Pt4+
(3)墹值还随电子给予体的原子半径减小而增大:
I<Br<Cl<S<F<O<N<C
墹值可用两部分贡献的乘积来表示:
墹=f(配位体的贡献)×g(中心离子的贡献)
墹值大的称为强场,墹值小的称为弱场,这是对同一种对称性的配位场而言的。对于不同对称性的配位场的情况,按照晶体场理论计算得出以下典型的关系式:
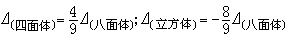
负号表明它们的两组轨道(或能级)的次序刚好颠倒。
依据构成原子和分子的电子层结构的基本原理,讨论弱场和强场下中心离子d电子的排布情况。例如,八面体配合物CoF咶、Fe(NH3)嵜和Co(NH3)扟、Fe(CH)忹的中心离子均为d6组态,在不同场强度的排布见图2。
 稳定化能
稳定化能
由图2 可以看出,由于配位场的存在,使得中心离子的d轨道能级分裂,大多数的d电子都趋向于处于较低的能级,体系较为稳定。这表明由于配位体的存在,中心离子的电荷分布不再是球形对称的,而是产生电偶极矩,这种电偶极矩与配位体的相互作用产生一种附加能量,称为配位场稳定化能,使得配合物稳定。由图1和图 2看出,一个电子处在 t2g轨道对体系稳定化能的贡献是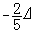 ,而处在eg轨道则贡献
,而处在eg轨道则贡献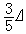 (5个d轨道权重平均值为零)。因此,对于八面体场
(5个d轨道权重平均值为零)。因此,对于八面体场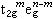 组态和四面体场的
组态和四面体场的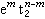 组态的配位场稳定化能(表2)用以下两个公式计算:
组态的配位场稳定化能(表2)用以下两个公式计算:

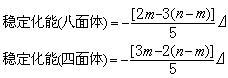
因此,在强场情况下电子趋向于在较低的能级,于是,中心离子为d4、d5、d6、d7者能形成较稳定的低自旋配合物,不过,自旋相反的孤对电子还有相斥作用,所以实际的稳定化能的计算值还应扣除这个称为成对能的数值。稳定化能一般由光谱测定或用点电荷模型作近似估计,利用稳定化能可进一步探讨络合物的热力学和反应动力学性质。
杨-特勒畸变如果一个非线形分子处于轨道简并态,则该分子要发生变形,使简并消除。这表明杨-特勒畸变引起附加的稳定化能使配合物稳定。有些配合物原来就不是严格的正多面体配位或者由于配位原子在平衡位置振动而偏离正多面体配位,产生静态扭歪。扭歪的八面体有两种构型,即沿四次轴拉长或缩短。例如,Cu2+(d9)的八面体配合物ML6,其中心离子的电子组态为t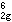 e
e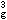 ,三个eg电子有
,三个eg电子有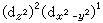 和
和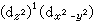 两种分布方式,前者表明z轴方向的电子密度比xy平面的x,y方向高,更有效地屏蔽核电荷与配位体的作用,因此,z轴方向配位体与中心离子的静电作用能较小,就产生向z轴方向拉长的畸变。同样对 (d
两种分布方式,前者表明z轴方向的电子密度比xy平面的x,y方向高,更有效地屏蔽核电荷与配位体的作用,因此,z轴方向配位体与中心离子的静电作用能较小,就产生向z轴方向拉长的畸变。同样对 (d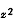 )1(d
)1(d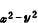 )2组态分析,则出现沿z 轴方向缩短的畸变。多面体产生各种畸变现象称为杨-特勒效应。
)2组态分析,则出现沿z 轴方向缩短的畸变。多面体产生各种畸变现象称为杨-特勒效应。
如果是中心离子和配位场之间有显著重叠的共价配合物,要更真实地反映其化学键性质,必须采用分子轨道理论。
对于ML6的八面体配合物, 中心离子的一个s、三个p和两个d原子轨道与6个配位体的σ轨道结合, 组成6个σ成键轨道和6个σ*反键轨道。过渡金属的d电子则处于t2g非键轨道或部分在弱反键e
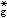 轨道上。另外,当配位体可能进一步提供p轨道,与中心离子t2g轨道形成π成键和π*反键轨道(图3)
轨道上。另外,当配位体可能进一步提供p轨道,与中心离子t2g轨道形成π成键和π*反键轨道(图3)
时,中心离子的s、p、d轨道和配位体的σ轨道都按八面体对称性分类为单重、双重和三重简并轨道a1g、 eg、t1u。相同对称性的中心离子的原子轨道和配位体σ轨道组合为分子轨道,其分子轨道组合形式和能级次序如图3所示。如果把配合物分子的价电子按以上能级图依序填入,可得配合物的电子组态。金属离子原子轨道和配位体轨道对其分子轨道组成的贡献不一定是等同的。如果配位体轨道贡献大,则占据该分子轨道的电子主要体现配位体的性质;反之,则体现金属离子的性质。
电荷迁移光谱如果上述分子轨道的电子产生跃迁,则出现电子光谱;电子由主要为配位体特性的分子轨道跃迁到主要为金属离子特性的分子轨道或相反情况,相当于电荷由配位体迁移到金属离子或相反情况,则出现电荷迁移光谱。例如,ML6配合物可能有三种电荷迁移光谱带的电子跃迁:
(1)配位体与金属离子只形成σ键,可能有σ→t2g和σ→e壛跃迁,可与金属还原带联系起来。
(2)配位体与金属离子形成σ和π键,可能有σ→t2g和σ→e壛以及π→t2g和π→e壛两类跃迁,也是相当于金属还原带。
(3)配位体有未占据的反键轨道π*,金属离子反馈电子到配位体,除了可能有σ→t2g和σ→e壛跃迁外,还有t2g→π*或e壛→π*跃迁,这相当于金属氧化带。
显然,分子轨道理论解释电荷迁移光谱是十分成功的。分子轨道理论在处理配合物结构和说明它的物理性质、化学性质上,比晶体场理论略高一筹。20世纪50年代以后,对配合物进行了大量的分子轨道理论计算,采用了非经验的自洽场从头算和xα方法以及半经验的全略微分重叠、间略微分重叠等量子化学计算方法。
随着无机和有机配合物合成的日益增多和各种结构与性能的研究,配位场理论不断发展,成为近代重要的化学键理论之一,是理论物理和理论化学的一个重要分支。它在解释配位化合物的结构与性能关系、催化反应机理,激光物质的工作原理以及晶体的物理性质等方面都得到广泛的应用。