[拼音]:xiantianxing daixie quexian
[外文]:inborn errors of metabolism
由于编码酶蛋白的结构基因发生突变而带来酶蛋白的结构异常;或者由于基因的调控系统异常而带来酶蛋白的量的变化,从而引起的先天性代谢紊乱。遗传方式一般都属常染色体隐性(见人类遗传性疾病)。
简史英国医学家A.E.加罗德在1902~1908年对尿黑酸尿症、胱氨酸尿症、白化症和戊糖尿症的患者进行了详细的研究,指出这些疾病都是由于某一代谢途径中的某一酶促反应发生遗传性障碍所造成,从而提出了先天性代谢缺陷概念。加罗得的假设在半个世纪以后才被证实。1952年美国学者G.T.科里发现糖原累积病Ⅰ型是由于患者肝脏内缺乏葡萄糖-6-磷酸酶而引起的;1955年日本学者九木田等证实了白化症患者毛囊基质内的黑色素细胞缺乏酪氨酸酶;1958年法国学者B.N.拉迪等证实了尿黑酸尿症患者肝脏内缺乏尿黑酸氧化酶,1970年Y.M.王等证实了戊糖尿症患者的红细胞内缺乏木糖醇脱氢酶。上述疾病都是中间代谢发生障碍的结果。
机理酶蛋白都由基因编码。如果编码某一种酶蛋白的基因发生的突变使所编码的蛋白质的活性中心发生了改变,那么这一种酶的活性往往降低甚至完全丧失,相应的生化反应也就不能顺利地进行,由此可以造成这一反应的底物或前体的积贮或者造成正常产物的不足或异常产物的出现。如果前体是有毒害的或正常产物是必需的,就会带来先天性代谢异常疾病。
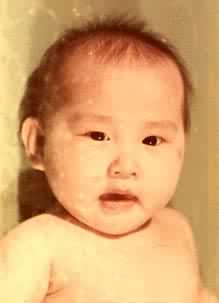
苯丙氨酸经苯丙氨酸羟化酶的催化成为酪氨酸(反应1)。 如果编码这一酶的基因发生突变而使这一反应不能顺利进行,那么体内苯丙氨酸积贮,尿中出现苯丙氨酸的氧化产物苯丙酮酸、苯乳酸、 苯乙酸和邻-羟苯丙酮酸等异常产物,从而导致苯丙酮尿症(见彩图);患者脑发育受损害,多数智力低下。由于正常产物酪氨酸又是黑色素的前体,所以酪氨酸的不足使患者毛发和肤色都较浅。
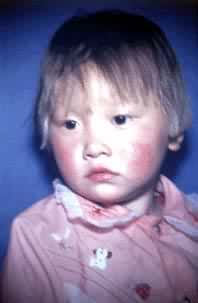
酪氨酸一方面经过许多生化反应而生成黑色素,另一方面经过两个生化反应而成为尿黑酸,后者又进一步分解成为延胡索酸和乙酰乙酸或转变成为一种褐黄色物质。如果基因突变使酪氨酸转氨酶失活,那么反应2便不能顺利进行,于是发生高酪氨酸血症(或称酪氨酸血症Ⅱ),患者血液中酪氨酸增多,智能低下。如果对-羟基苯丙酮酸氧化酶失活则反应3不能顺利进行,出现遗传性酪氨酸血症(或称酪氨酸血症Ⅰ),患者肝脏肿大,肾功能受损。如果尿黑酸氧化酶失活则反应 4不能顺利进行,发生尿黑酸尿症,患者的尿液在接触空气后变为黑色,老年时容易由于褐黄色物质在软骨组织中的沉积而引起关节炎。如果酪氨酸酶失活则反应 5不能顺利进行,黑色素不能形成,于是导致白化症(见彩图)。
 类型
类型
目前已知先天性代谢缺陷所带来的疾病共有1000余种,从不同的角度可以区分为多种类型:
(1)根据代谢物的生化性质,可以分为碳水化合物、氨基酸、脂类(包括鞘磷脂、糖鞘脂、糖脑苷脂、神经节苷脂、粘脂)、粘多糖、类固醇激素、核酸(包括嘌呤和嘧啶)、金属离子、维生素等代谢缺陷。
(2)根据疾病的发病机理,可以分为主动转运障碍(例如肾性糖尿病、尿崩症所造成的缺陷)、代谢物累积或积聚过多(例如糖原累积病、粘多糖病所造成的缺陷)、中间代谢障碍(例如尿素循环障碍、卟淋病)以及合成障碍(例如高脂蛋白血症、肾上腺皮质类固醇合成障碍所造成的缺陷)。
(3)根据遗传方式,可以分为单基因和多基因两大类。单基因又可分为常染色体隐性、常染色体显性、X连锁隐性和X连锁显性等类别。
先天性代谢缺陷在临床上可以是无症状的,例如戊糖尿症;可以是在一定外界因素诱发下才出现症状的,例如葡萄糖-6-磷酸脱氨酶(G6PD)缺陷(见药物遗传学);也可以是不经诱发便呈现持续症状的,其中轻度的可以长期存活,甚至达正常存活年龄,严重的往往导致死亡,例如脂类累积病的患儿一般都在婴儿期死亡。
预防和治疗从遗传方面减少群体中先天性代谢缺陷发病率的措施主要是:
(1)避免近亲结婚。
(2)遗传咨询。根据家系中病员的分布,疾病的遗传方式,可推算生出有病儿童的几率,就可决定应否生育或继续妊娠等。
(3)杂合体检出。隐性遗传的杂合体外表正常,然而相应的酶的活性常界于正常人与隐性纯合体患者之间。因此可通过酶活性的测定检出杂合体,但如果测定的结果与正常值重叠,就须用其他方法,例如采用苯丙氨酸耐量试验可以检出苯丙酮尿症杂合体。在高发地区的群体中检出杂合体可以有效地预防先天性代谢缺陷病的发生。
(4)产前诊断。某些先天性代谢缺陷可以通过测定培养的羊水中胎儿脱屑细胞的酶活性而检出。到1979年为止,已经有75种先天性代谢缺陷可用此法检出。某些代谢缺陷例如杜氏肌营养不良或称假肥大性营养不良可以通过胎儿内窥镜抽取胎儿血标本检出。自80年代起已应用重组DNA技术产前诊断先天性代谢缺陷,从而进入基因诊断水平。这对于指导人工流产有很大的帮助,特别是对于已生育过先天性代谢缺陷患儿的孕妇更有意义。
先天性代谢缺陷的治疗主要采用酶疗法。70年代以来用在体内能降解的尼龙薄膜包装酶制成微囊(人工细胞),把这种人工细胞定期给患者输入是治疗酶缺陷很有前途的方法。补充缺乏的代谢(产)物,避免接触诱发物质,诱导代谢以减少体内积贮的物质,使用抑制剂,使用辅酶,限制摄入缺陷酶的底物以及补充缺乏的物质等都是临床上常用的疗法。
在病毒和噬菌体以外的任何生物中都可以发生先天性代谢缺陷。各类营养缺陷型便是由于在氨基酸、核苷酸或维生素等的代谢过程中出现的先天性代谢缺陷的结果。营养缺陷型是遗传学研究,特别是微生物遗传学研究的重要工具。
- 参考书目
- D.J.H.Brock,D.Mayo,The Biochemical Genetics of Man,Academic Press,London,1978.