[拼音]:haiyang wuli huaxue
[外文]:marine physical chemistry
海洋化学的理论核心。它应用物理化学的理论、观点和方法,来研究海洋中的化学问题和地球化学过程,包括海水、悬浮粒子和沉积物、微表层海水和沉积物间隙水等海洋体系的组成、物理化学性质和结构、海洋及其环境(大气、洋底、河口、洋底火山等)所组成的体系中发生的一切物理化学过程。
1959年在纽约召开的国际海洋会议上,L.G.西伦作了题为“海水物理化学”的演讲。以后,“海水物理化学”这一名称便被广泛采用。海洋物理化学的发展初期,研究对象主要是海水、嗣后研究范围逐渐推广到海底沉积物和海洋的其他界面上,就发展成了海洋物理化学。
海洋物理化学的先驱性工作在20世纪初已有报道。1901~1908年,M.H.C.克努曾和V.W.埃克曼建立了经典的海水状态方程式,它是联系海水的密度、温度、盐度和压力等的一种数学表达式。二氧化碳-碳酸盐体系,是海洋中控制海水的pH值、决定海洋沉积物的沉积过程等的复杂的平衡体系,虽已研究了几十年,但直到20世纪50年代末才初具雏型。60年代初,西伦、R.M.加勒尔斯等人把化学平衡理论等物理化学原理,严格、系统和定量地应用到海洋中,研究海水中元素的存在形式,建立海水化学模型,为海洋物理化学的建立和发展奠定了基础。在这以后的一段时间内,海水化学模型(包括微量元素的溶存形式)的研究,成了海洋化学中众所瞩目的一个研究内容。自60年代以来,人们又把电化学、化学动力学、胶体和表面化学、量子和统计化学的理论和实验方法应用到海洋中,不仅研究了海洋水体,而且涉及海洋悬浮体、海水微表层、海洋沉积物等,使海洋物理化学具有如下一些特点:
(1)所研究的海水体系是地球上极其复杂的化学平衡体系之一(图1)。
(2)所研究的化学过程,许多是陆地上少见的低浓底(1ppm以下)化学过程。
(3)视海水为多组分电介质溶液,其活度系数理论已有较深入的研究。
(4)海水是具有胶体化学特性的水溶液,其中存在着一定量的固体悬浮粒子,海水中“微量元素-有机物质-固体粒子”的相互作用及其液-固分配理论的研究,是现时海洋化学研究的新动向之一。
(5)在海洋中进行的化学反应,压力范围很宽,可自 1个大气压至1000大气压左右,因此必须考虑化学过程的压力效应。
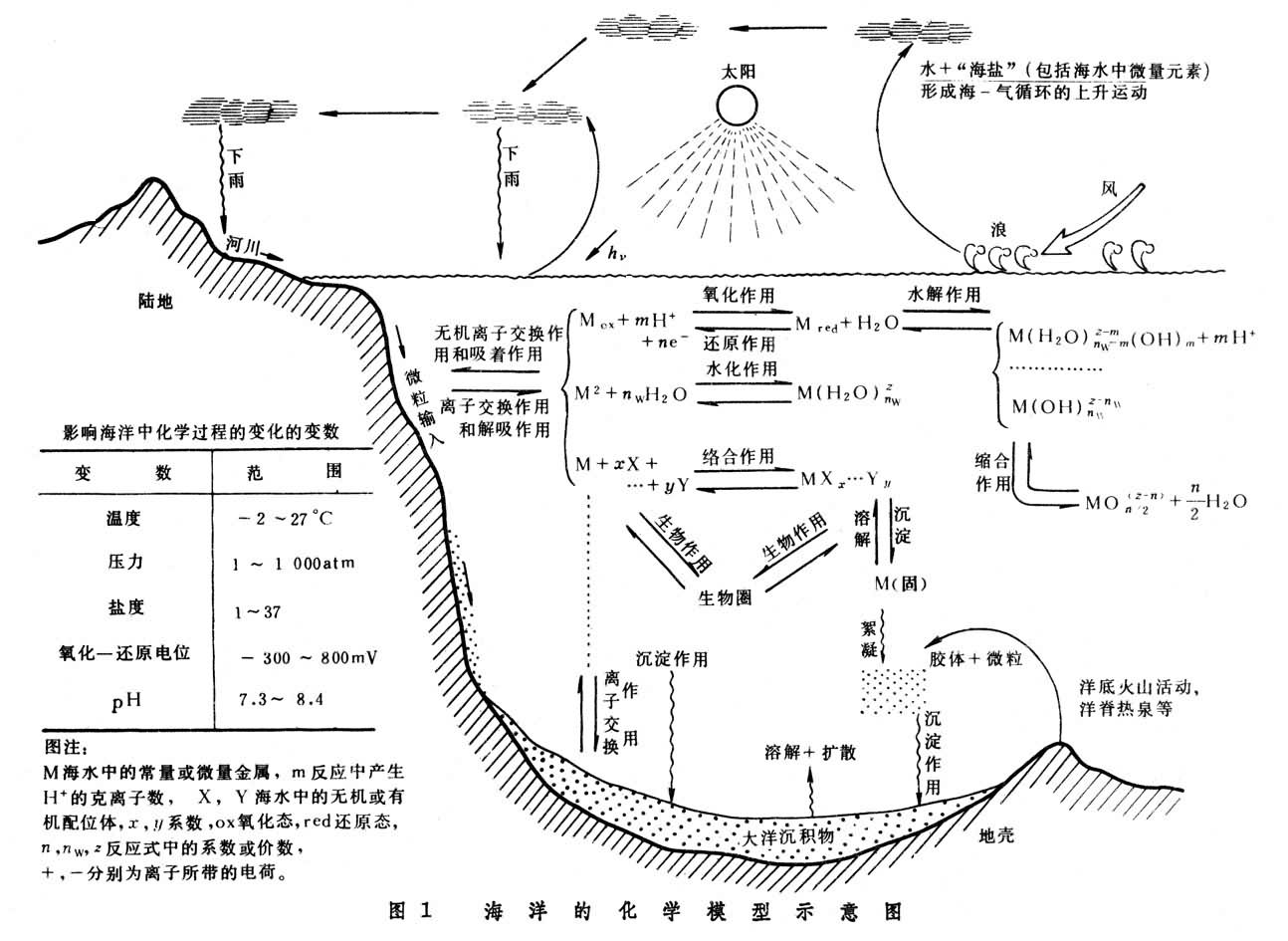
海洋物理化学的基本内容包括:海水活度系数,海水化学模型,海水中微量元素在固体粒子上液-固分配的理论,压力和温度对海洋中化学平衡的影响,海水的pE和pH图,海洋中化学过程的动力学研究,海洋中元素的逗留时间和海洋化学微观研究等。
海水各组分之间存在着错综复杂的相互关系,如果把海水划成“海盐”和水两种组分,则主要考虑下述3种关系:
(1)水分子之间的相互作用,即液态水的结构问题。液态水理论众说纷纭,例如扭变氢键模型(连续体理论),它假设冰变水时,氢键变得更加易被弯曲、扭转和变型,但尚未拆开,氢键扭变可用经典统计方法处理;笼合体模型,它假设液态水成为一种自笼合体,即是许多由带氢键分子形式的五边形十二面体笼子组成的网络,笼子空腔中包藏着水分子,但它们不与笼结构键合;闪动簇团模型,它将液态水视为氢键结合的水分子的“闪动簇团”在略为“自由”的水中游泳的混合体,这一模型与分配函数联系起来,可用统计力学方法计算热力学性质,经再三改进后,计算得出的自由能、内能、熵和热容之值,与实测值基本一致。后面两种理论均属混合型模型理论。
(2)海水中“海盐”离子与水的相互作用,即离子水化作用。其结果将对离子的电导和温度系数、热力学性质和动力学性质、海水中元素的存在形式及其稳定性等都有不同程度影响。
(3)海水中“海盐”离子之间的相互作用。可用海水活度系数和海水渗透系数加以表达。这三种关系中,最重要的特征量是活度系数。
海水活度系数与一般的水溶液或天然水(河、湖、泉、雨之水)相比,海水的一个重要特征是含有众多的盐类。例如,大洋水的盐度约为S=35,结果使海水与盐度极低的无限稀溶液之间的物理-化学性质有很大的差异。为了表达两者的这种差异程度,在海洋物理化学中使用了海水活度系数(主要用于溶质)和海水渗透系数(主要用于溶剂)。
计算海水组分平均活度系数和单独离子活度系数的理论模型和公式很多,有离子缔合理论、特殊相互作用模型、斯卡特查尔德公式、水化模型、静电模型(包括戴维斯公式)、簇积分展开理论、皮策公式等。虽然使用离子选择电极可求得单独离子的活度系数,但其精确值的测定和理论计算,特别是对海水这样的高盐度体系,尚未彻底解决。在实际工作中,可用特殊相互作用公式,如斯卡特查尔德公式和皮策公式等计算海水的活度系数。总结起来,上述各计算公式可用下述公式概括之:  式中德拜-休克尔项是指德拜-休克尔理论的极限定律计算所得之值。在上述诸理论中,以簇积分展开理论和皮策公式最优,计算结果与实测值之差在实验误差范围之内。皮策公式不仅可计算海水中常量组分离子的活度系数,而且在皮策公式问世后不久即被海洋化学家应用于海水中微量组分离子活度系数的计算,结果与用特殊相互作用模型计算的更加一致。1982年,此式已成功地应用于死海水(离子强度(±)埄10)中主要离子的活度系数的计算上。皮策公式的提出,使得具体计算活度系数的工作大体上得到解决。
式中德拜-休克尔项是指德拜-休克尔理论的极限定律计算所得之值。在上述诸理论中,以簇积分展开理论和皮策公式最优,计算结果与实测值之差在实验误差范围之内。皮策公式不仅可计算海水中常量组分离子的活度系数,而且在皮策公式问世后不久即被海洋化学家应用于海水中微量组分离子活度系数的计算,结果与用特殊相互作用模型计算的更加一致。1982年,此式已成功地应用于死海水(离子强度(±)埄10)中主要离子的活度系数的计算上。皮策公式的提出,使得具体计算活度系数的工作大体上得到解决。
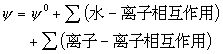
对任一海水物理化学性质ψ而言,考虑到上述3种海水组分之间的相互作用后,可用下述公式表达:
 海水化学模型
海水化学模型
研究海水中元素的存在形式。元素的存在形式是元素海洋地球化学的一个研究内容,又是影响海水中元素迁移变化规律的一个重要因素,因此海水化学模型是元素海洋地球化学的一个重要组成部分。它对海水分析、海水中微量元素提取机理、海水中微量元素与悬浮粒子相互作用等,均是常常需要考虑的前提。
1962年,加勒尔斯和M.E.汤普森为研究海水中常量元素的溶存形式,提出了离子对模型。后来这种海水化学模型的应用范围已不限于常量元素,而且推广到微量元素和有机物质,并提出络合物模型、HSAB(硬软酸碱)模型、活度系数模型和微观结构参数模型等。这些化学模型大致可分为3类:
(1)用活度系数和渗透系数来描述离子之间的相互作用(例如M.惠特菲尔德用皮策公式),由此推断海水及其组分的若干性质。但这类方法缺乏一般海洋化学工作者所习惯的形象化的结构图式。
(2)离子对模型或络合物模型。西伦、加勒尔斯和汤普森等首先提出。在已知海水化学组成、络合物的稳定常数和活度系数的基础上,应用化学平衡理论进行计算,可得常量元素和微量元素的化学模型(表1)。

(3)微观结构参数模型,是由张正斌、刘莲生、陈镇东提出的。由海水中微量元素的化学模型与结构参数 的关系(图2)可见属软酸和中间酸的元素主要形成M-Cl型络合物,属硬酸的元素则主要形成M-OH型络合物。CA/EA值在0.1左右的元素的溶存形式比较复杂,M-Cl,M-OH,M-CO3,M-SO4等形式络离子都可能存在。在河口区,元素的溶存形式原则上也可用上述方法计算,但具体运算时需注意到河口水体的成分、pH、氧化-还原条件、离子强度、有机物的成分和含量、悬浮物质的种类和含量等因素的影响。
的关系(图2)可见属软酸和中间酸的元素主要形成M-Cl型络合物,属硬酸的元素则主要形成M-OH型络合物。CA/EA值在0.1左右的元素的溶存形式比较复杂,M-Cl,M-OH,M-CO3,M-SO4等形式络离子都可能存在。在河口区,元素的溶存形式原则上也可用上述方法计算,但具体运算时需注意到河口水体的成分、pH、氧化-还原条件、离子强度、有机物的成分和含量、悬浮物质的种类和含量等因素的影响。
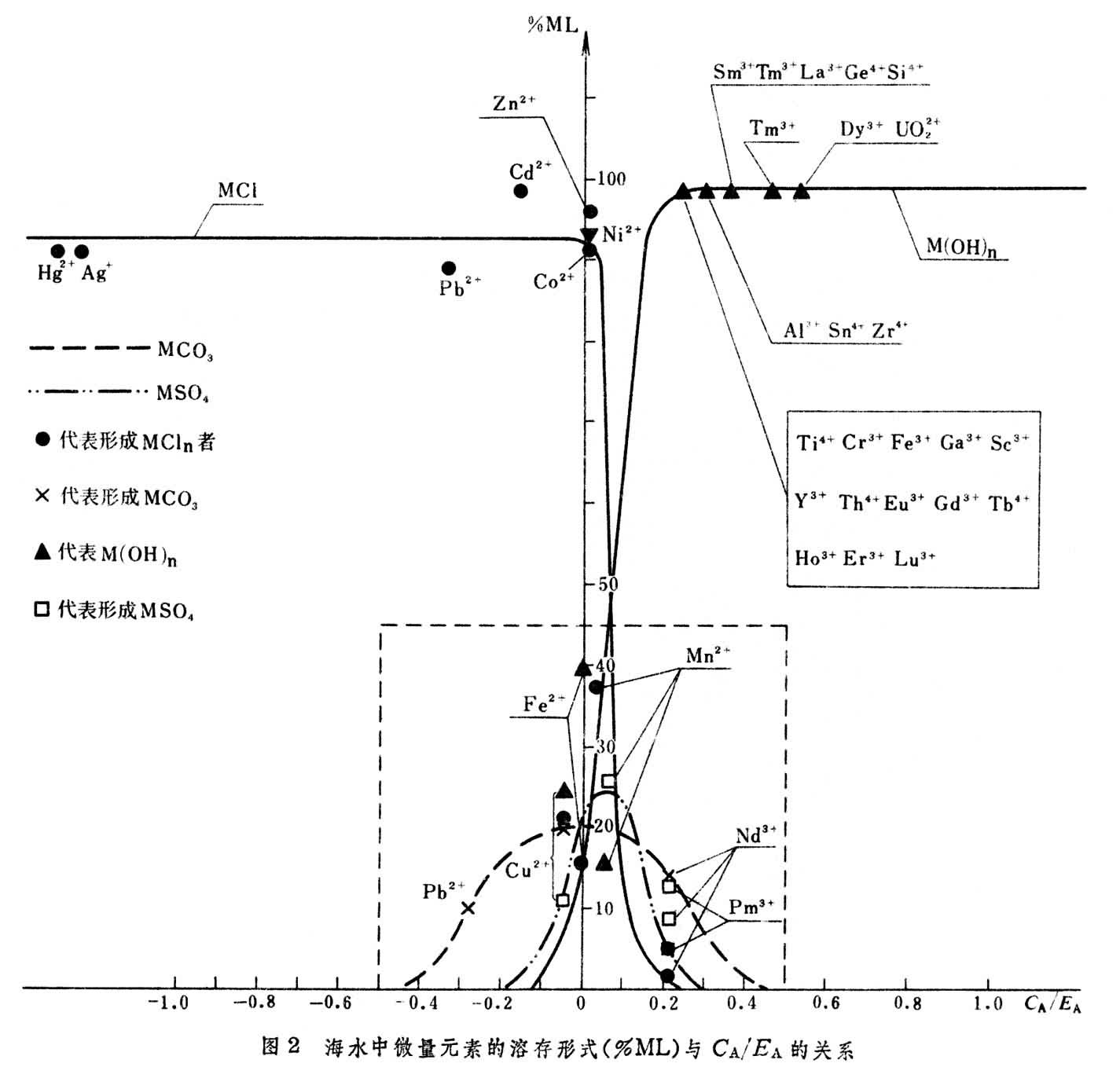
不同学者提出的海水常量组分的化学模型将近十种,结论大体类似,但对“M-Cl”型离子对是否生成这一点,有两种相反的见解,多数模型不考虑“M-Cl”型离子对生成,但一些实验和理论计算认为不仅要考虑“M-Cl”型离子对的生成,而且其生成率(%)可能比“M-SO42-ν”高些。这两种见解尚未统一。在海水中微量元素的存在形式方面,也存在着不同模型结论不同的情况。例如文献上一般报道的U(Ⅵ)在海水中的溶存形式为UO2(CO3)嬈3,但中国一些学者经实验和理论计算证明了主要的存在形式为UO2(OH)婣,UO2(CO3)嬈3为次。此外,目前有机络合物的重要性日渐重要,例如铜、锌等的有机存在形式占总分配50%以上,考虑了有机络合物生成的海水化学模型的研究,已列入研究日程。
海洋中化学元素与固体粒子的相互作用海水含有大量固体粒子,具有胶体化学或表面化学特征。这些固体粒子上微量元素的液-固分配,遵循着低浓度的界面化学和胶体化学的规律。这方面的研究是探讨海洋中元素迁移变化规律的理论基础,是河口化学的重要内容,也是海洋污染和防污、海水化学资源提取的基础研究。这方面研究虽然多数针对着微量元素,但常量元素在海水-洋底界面间的离子交换作用,也属于这种作用范围,例如碳酸盐沉积物的钙-镁交换
Mg2++2CaCO3匑Ca2++MgCa(CO3)2
又如对海水pH的形成可能有重要作用的K+-H+交换:
3Al2Si2O5(OH)4(固)+2K+匑2KAl3Si3O10(OH)2(固)+3H2O
海水中元素与固体粒子相互作用的理论主要有3种:
(1)R.O.詹姆斯和T.W.希利等提出的化学吸附理论;
(2)W.施图姆、P.W.欣德勒、C.W.戴维斯、J.O.莱基等提出的表面络合理论;
(3)张正斌、刘莲生提出的表面分级离子(配位子)交换理论。三者各有所长,都尚在发展中。在具体研究中,进行得最早和最多的是离子交换率(或吸着率)和pH的关系。研究得最多的无机离子交换剂是水合氧化物,其次是粘土矿物、氟石等,涉及的微量元素20多种。由离子交换率-pH关系研究得到的图形呈S型或反S型,可推断作用过程为阳离子交换还是阴离子交换;由曲线的位置和范围大小可推断相互作用的强弱和交换离子的价数。
海水中微量元素在固体粒子上的作用等温线,常见报道的有:
(1)用相应的弗罗因德利希经验公式表达,即ε=ɑ(M)b。式中ε为交换量(或吸着量),(M)为溶液中元素的平衡浓度,ɑ和b为系数。石桥雅义等证明海水中微量元素(20个左右)在水合氧化铁上的作用遵循此式。这是海洋化学中最流行的等温式。
(2)用相应的朗缪尔公式表达。例如Zn、Pb、Cu、Cd在SiO2和γ-Al2O3上的作用,Co、Zn、Ca、Na在δ-MnO2上的作用等。此式不如弗罗因德利希公式用得普遍。
(3)用兼具弗罗因德利希和朗缪尔两式特点的公式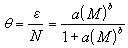 表达(式中N 为最大交换量)。可得与实验很好相符合的结果。上述三式都只能解释简单的等温线。为了推广应用到比较复杂的等温式的场合,上式又改进为
表达(式中N 为最大交换量)。可得与实验很好相符合的结果。上述三式都只能解释简单的等温线。为了推广应用到比较复杂的等温式的场合,上式又改进为
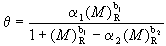
式中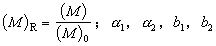 为可调参数。
为可调参数。
(4)用固体表面分级离子交换理论的公式
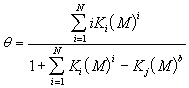
表达。式中Ki是i级离子交换产物的生成常数,Kj是生成不溶物的生成常数。这是一种普遍的等温式。它不仅可表达海洋化学上已有的等温线,而且可以表达所测得的“台阶式”的和“离子交换-沉积式”的新型等温线。
海洋固体粒子主要由粘土矿物、金属氧化物和有机物质三者组成。此三者分别与微量元素的作用均有大量的实验结果报道,但三者中何种成分对金属的交换起决定性作用,则各家说法不一。一般认为粘土矿物含量百分比最大,本身对微量元素虽有离子交换作用,但交换量不大,主要起“载体”的作用;水合氧化物含量百分比虽小,但离子交换量较大,一些学者认为它可能是与微量元素作用的主角;有机物一般与微量元素有强的络合(或螯合)作用。在有机物同时易与固体粒子结合的情况下,则它可能是海水中微量元素与固体粒子作用中的关键物质。一些学者提出海水中存在的固体粒子可分别由无机物和有机物所组成,相应地建立了无机物模型和有机物模型,再同上述元素与固体粒子相互作用的理论及元素在海洋中的逗留时间相结合,经定量处理后,可得出海水中有机物起主要作用的结论。然而,有机物所起作用的本质和定量规律,尚待进一步研究。
关于海水中微量元素与固体粒子相互作用的动力学研究,就海水中铀(Ⅵ)在水合氧化钛上的提取机理来说,有一种看法为:
(1)可用“扩散-表面吸着和转形-化学反应-解吸-扩散”的五步机理表达;
(2)在天然海水条件下,过程速率由液膜扩散所控制;实验测得过程活化能Ea为8.5(千卡/克分子)左右,适与其他文献报道的扩散活化能接近;
(3)通水过程的pH研究和红外光谱研究皆说明,机理中“化学反应”一步是阳离子交换或者“络合缩水”反应,反应级数n=1。
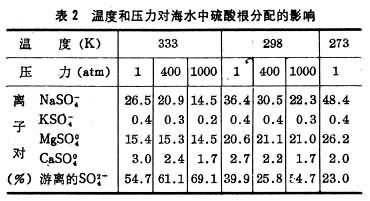
温度和压力对海洋中化学平衡的影响海洋往往被人们形容为一个恒温箱,在不同的季节,温度的垂直变化很少超过30°C,这是生命可能起源于海洋的一个重要条件。但温度的这种周期性变化,已足使海水的一些物理-化学性质,例如溶解氧等发生显著的变化。另一方面,海洋作为化学环境的又一特征,是静压力。从表面的1大气压到最深洋底的1000大气压左右,它影响着海洋中各种化学平衡。温度和压力不仅对海水的活度系数、海水中元素的存在形式及其与固体粒子的作用,有不可忽视的影响,而且对海洋中的氧化-还原作用,海水扩散系数、溶度积等,都有一定的影响。温度和压力对海水中硫酸根分配的影响,就是一个例子(表2)。
温度和压力对平衡常数的影响可用下式估算:
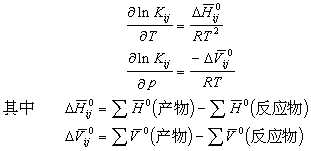
由此可见有两种研究压力对平衡影响的方法,①偏克分子体积法,由产物和反应物的偏克分子体积计算-Δ堸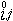 ;
;
(2)平衡常数法,即直接通过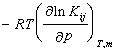 的测量而求得之。
的测量而求得之。
对海水中的主要盐类和离子,不仅已测得比较精确的实验值,而且在理论上一些学者分别提出了不同的定量计算方法和公式,得到了与实验值比较一致的结果。再通过上述公式,即可估计出压力对所研究体系的影响。
海洋中的氧化—还原作用海水中元素的化学模型,除了与海水中的无机配位体、有机配位体和海水的pH三因素密切相关外,还被海水体系的氧化 -还原环境所决定,作为体系氧化-还原能力的量度,海洋化学上使用pE,它被定义为:pE=-log{e}。e代表电子,{e}代表活度,即pE为电子活度的负对数。表层海水pE的理论最高值为12.5±0.2,这是假设体系达到氧化-还原平衡的理论值。实际海水情况决非如此简单,各家见解不一,尚待探讨。
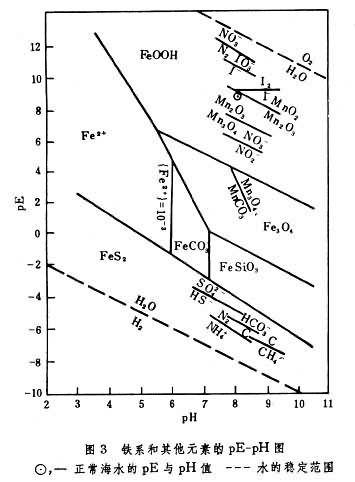
pH和pE都对海水中元素的溶存形式有显著影响,故用海洋中元素的pE-pH图,可以研究元素的存在形式(图3),由此可判断在具体研究的海水条件下,元素处于氧化-还原状态。海水中氧化-还原态变化的元素主要有H、O、C、N、Fe、Mn、S、Cr、I等,它们在各种状态(含氧、缺氧等状态)的海水中存在的形式,对海洋生物和海洋地质等有重要的参考价值。
海水中金属和合金的电化学位是研究海洋环境中金属腐蚀和保护的一个最基本的物理-化学量(见海水中金属的腐蚀),其中纯金属的电化学位有明显的规律性。例如可把它们分成 3类:
(1)Zr、Ta、Nb、Ti、Pt、Pd、Mo、V、W;
(2)Au、Ag、Cu、Fe、Cd、Zn、Mg;
(3)Ni、Bi、In、Pb、Sn、Al。通过设计两套哈伯-玻恩循环可以证明:这与它们在海水中生成的络合物MLn中n=0,1,2的情况是一一对应的(其中L为Cl-、OH-、SO娺-、CO卲-等)。
海洋中化学过程的动力学研究这方面的研究是比较复杂的。例如:存在于海水中的多种配位体和络合物,对金属在海洋中的腐蚀反应及磷酸盐、硅酸盐、碳酸盐等的沉淀溶解反应的过程和速率,都有明显的影响。此外,海水中固体的成核作用,晶体的生长,物质的水化和脱水作用,无机物和有机物的氧化-还原作用,吸着和解吸作用等化学反应的速率,也往往在一定条件下对某些化学过程有关键性的影响。还有一些化学过程,例如某一无机物、有机物或核素的反应过程,同时受到大洋表层流、深层环流、大洋湍流,或者近岸海域和河口海湾的潮流、密度流和港湾环流等的显著影响。因此,成功地研究现场海洋中化学过程动力学的范例很少。然而海洋中一些重要体系,例如碳酸盐体系中的一些重要反应的速率和机理,已有比较深入的研究。包括:
(1)CO2在海水中溶解和垂直迁移。其中起控制速率作用的是 CO2穿过界面和 CO2的水化和脱水反应 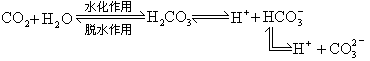 其中第一步的速率很小,故成为控制整个过程的速率的关键,其他均相化学反应则均较快,关键反应的反应级数和反应速率常数已经测定。
其中第一步的速率很小,故成为控制整个过程的速率的关键,其他均相化学反应则均较快,关键反应的反应级数和反应速率常数已经测定。
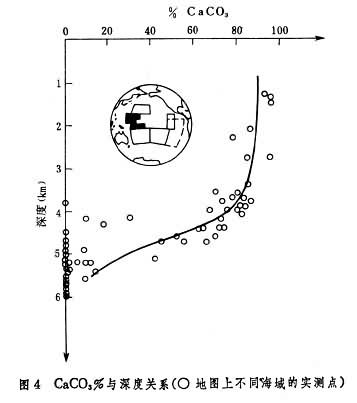
(2)碳酸钙的沉淀和溶解。对 CaCO3而言世界大洋上层大部分是饱和或过饱和的,而下层往往是不饱和的。海水恰好饱和的深度叫饱和深度。在饱和深度以下2000米或更深处,沉积物中CaCO3的丰度锐减。此外的深度称为补偿深度(图4),它受动力学因素所决定。
 海水中元素的逗留时间
海水中元素的逗留时间
海水中某一元素的现有总量A,除以单位时间内河水补充的量ΔQ或沉淀为沉积物的量ΔQ′,其商称为该元素在海水中的逗留时间τ ,即
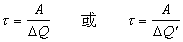
这一定义基于如下的简化假设:
(1)就化学组成而言,世界海洋可视为一稳恒体系,故单位时间内输入的物质的量,相当于析出沉积物的量。
(2)进入海洋中的元素达到和海水完全混合的时间与逗留时间相比是短暂的。1952年,T.F.W.巴尔特利用河水流量,E.D.戈德堡和G.O.S.阿伦尼乌斯等曾分别于1958年和1975年利用向海底沉淀的沉积物量,研究和计算了逗留时间。这两种方法的结果大体一致。
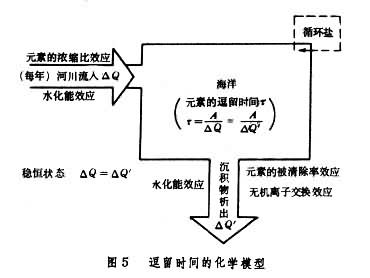
利用每种元素在海洋中逗留的平均时间τ ,可研究该种元素在海洋中的相对活泼性和它们的环境性质,进而解释海洋中发生的重要无机反应。τ 也是研究海底矿物(例如太平洋底锰结核等锰铁矿物等等)的形成和组成的一个重要的海洋物理化学参数。元素海洋地球化学的重要内容之一,是研究海水中元素含量与河水中元素含量的比值(浓缩比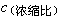 ) 和洋底沉积物中元素含量与海水中元素含量之比值(被清除率
) 和洋底沉积物中元素含量与海水中元素含量之比值(被清除率 )。它们与τ的关系可通过图5表达,并可导出下述关系式:
)。它们与τ的关系可通过图5表达,并可导出下述关系式:
τ=K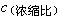
τ=K′/
上述公式表明:海洋中元素的平均逗留时间τ 与该元素的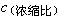 成正比;τ与该元素的
成正比;τ与该元素的 成反比,其中元素比例常数K或K′与水化能等海洋化学参数有关。用上述公式计算的τ 值,与巴尔特、戈德堡和阿伦尼乌斯的τ 值一致。
成反比,其中元素比例常数K或K′与水化能等海洋化学参数有关。用上述公式计算的τ 值,与巴尔特、戈德堡和阿伦尼乌斯的τ 值一致。
海洋化学微观研究
用组成海洋物质的原子与分子的结构观点,用量子化学的理论与方法,运用物质的某些结构参数,或通过大量实验结果和自然现象的归纳,或通过理论上的演绎,从定性和定量两方面来研究海洋化学的问题。
海洋化学的微观研究,大体上可分成两类:
(1)定性研究。例如通过“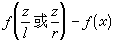 图”
图” ,
, 对海洋化学现象或海水物理-化学性质进行分类。又如通过“某一物理化学性质
对海洋化学现象或海水物理-化学性质进行分类。又如通过“某一物理化学性质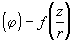 或
或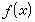 图”
图”
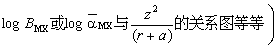 ,对φ 的规律性进行研究。
,对φ 的规律性进行研究。
(2)定量研究。即通过诸如
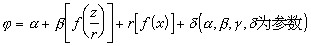
定量计算φ值。例如:海水中各种元素在水合氧化铁上的无机离子交换作用、氯化钾在各种无机离子交换剂上的作用、海水和河水中元素含量的浓缩比效应、海水中元素的清除作用、海水中主要盐类和离子的偏克分子体积、海水中纯金属的电化学位、离子的水化能、氢氧化物的溶解度、海水中元素的逗留时间、海水中的直线自由能关系、酸碱硬软度的定量标度、海水化学模型的微观定量计算等,张正斌、刘莲生等提出均可用上述形式的公式计算出相应的φ 值。
海洋化学正处于由定性描述向定量推理,由宏观向微观,由可逆平衡态热力学方法向不可逆、非平衡态、统计化学方法的方向发展的变革之中,它已经初步建立了自己的理论体系和实验方法,成为海洋科学中的一门严格而系统的分支学科。20多年来海洋物理化学的发展,对海水化学资源开发,海洋污染和防污染,海水腐蚀和防腐蚀等实验性较强的分支的发展,也起到积极的推动作用。