[拼音]:cuihua lilun
[外文]:theories of catalysis
对催化机理的理论解释。
中间化合物理论
认为催化剂先与某一反应物作用,生成活性中间化合物,此中间化合物再进一步反应而生成产物。1806年,法国的N.克莱芒等在研究一氧化氮对二氧化硫氧化的催化作用时,推测一氧化氮先与空气中的氧作用,生成某种活泼的中间化合物,然后该中间化合物使二氧化硫氧化,而本身又复原成一氧化氮。1835年瑞典的J.J.贝采利乌斯认为此反应的中间化合物是三氧化二氮。1851年英国的A.W.威廉森认为,在硫酸的催化下,乙醇的脱水反应也是先形成乙基硫酸,然后分解的过程,从此确立了中间化合物的学说。1930年英国的C.N.欣谢尔伍德等以碘蒸气为催化剂,进行乙醛蒸气的加热分解实验,发现均相催化的反应速率与催化剂的浓度成正比,而这个浓度在反应前后保持不变。这也说明催化剂先和某一反应物作用,生成活性中间化合物,后者又进一步反应,转变为产物,同时催化剂复原。这样便进一步证实和完善了催化中间化合物理论。此理论指出:
(1)催化剂直接参与化学反应;
(2)均相催化的反应速率与催化剂浓度的某次方成正比。实验证明该理论是正确的。
催化活化过渡态理论
认为反应分子经碰撞、接触后经过一个活化过渡态(形成活化络合物),再转变为产物。该理论于1932年由E.维格纳等人提出,后经H.艾林和M.波拉尼等人发展和完善。1940年此理论首先被K.J.莱德勒等应用于多相催化剂表面上的吸附、反应和解吸。1925年美国的H.S.泰勒提出固体催化剂表面只有一小部分能起催化作用,这部分称为活性中心。催化活化过渡态理论认为:反应分子先与催化剂表面的活性中心形成吸附活化络合物,然后反应,转变为产物,可用下式表示:
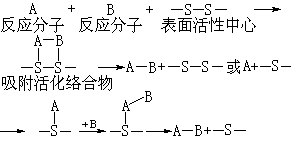
由于表面活性中心的影响,被吸附的分子变形,化学键松弛,同时形成活泼的吸附活化络合物,降低反应活化能,促进反应。此理论大致指出催化剂参与反应的本质,在催化反应中不需要一定形成较稳定的中间化合物。
多位催化理论
认为催化活化中常有多个活性中心对反应分子发生影响。此理论1929年由苏联的A.A.巴兰金提出。如果催化剂表面活性中心的空间结构(分布和间距)与反应分子将发生变化的那部分结构呈几何对应关系时,则被吸附的分子容易变形活化,即旧键较易松弛,新键较易形成。这称为几何对应原理。例如。环己烷分子可以在具有合适晶格常数(2.47~2.8埃)的面心立方或六方晶格的金属(如镍、钴、铁、钌、铑、钯、铂、铱、锇)上面进行六位吸附和脱氢。又如A-B及C-D分子被催化剂表面两个活性中心(用K表示)活化吸附,并相互作用,形成A-C及B-D分子而解吸的过程可示意如下:
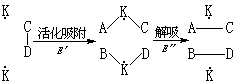
此理论还根据反应物及生成物的键能DA-B、DC-D、DA-C、DB-D及催化剂上吸附键的键能 DK-A、DK-C、DK-B、DK-D计算活化吸附所需能量 E′及解吸所需能量E″。当二者接近时,催化反应所需活化能最低。这称为能量对应原理。此理论的优点是提出了多位吸附及结构匹配和能量匹配的规律,缺点是模型过于简化。
活性集团催化理论
认为活性中心是催化剂表面上非晶相中几个原子组成的集团。此理论1939年由苏联的Н.И.科巴捷夫提出。此理论并认为活性集团中原子的数目与反应机理有关。例如铁高度分散在碳的表面时,可形成Fe2、Fe3等集团。对于合成氨反应,实验证明Fe3集团最为活泼。此理论所指出的集团概念及其与机理的关系是有启发意义的。
半导体催化电子理论
把催化作用描述为反应分子与催化剂表面之间的一种电子传递过程,而担负此传递任务的是用作催化剂的半导体(过渡金属氧化物)导带中的电子或满带中的空穴。苏联的Ф.Ф.沃尔肯斯坦于20世纪50年代提出该理论,并于1960年发表了专著《半导体催化的电子理论》。此理论的特点是把催化剂表面吸附的反应分子也看成是整个半导体的施主或受主,也就是把催化反应的两个方面:反应分子和催化剂(包括助催化剂)看成一个统一的整体,并且按照半导体的术语,把催化反应分成 n型反应(其反应的控制步骤受电子加速的反应)和 p型反应(其反应的控制步骤受空穴加速的反应)。n型半导体催化剂加速n型反应,p型半导体催化剂加速p型反应。例如,当反应分子的电离势大于催化剂的电子逸出功时,在极端情况下此反应分子将以负离子的形式吸附在表面上,这相当于增加了受主浓度,使电子逸出功上升。若这一吸附过程为反应的控制步骤,则此反应属n型反应,受电子加速,电子逸出功愈小的n型半导体催化剂,其活性愈高。催化剂中若添加施主杂质(金属价数大于催化剂的离子),将起助催化作用。
半导体催化电子理论过分强调了“自由电子”的作用,以及催化剂的活性与其宏观的电子迁移性能(如电导、电子逸出功等)之间的形式上的平行或反平行关系,因而局限性较大。到20世纪60年代后期,催化作用的模型已朝着化学键变化的微观模型发展,建立在现代化的化学键理论和活性中心的电子结构理论的基础上。