[拼音]:peiweijian
[外文]:coordination bond
一种共价键,成键的两原子间共享的两个电子不是由两原子各提供一个,而是来自一个原子。例如,氨和三氟化硼可以形成配位化合物:
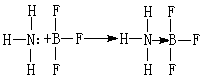
式中→表示配位键。在N和B之间的一对电子来自N原子上的孤对电子。
配位键是极性键,电子总是偏向一方,根据极性的强弱,或接近离子键,或接近极性共价键。在一些配合物中,除配体向受体提供电子形成普通配位键外,受体的电子也向配体转移形成反馈配键,例如Ni(CO)4中CO中碳上的孤对电子向镍原子配位形成σ配位键,镍原子的d电子则反过来流向CO的空π*反键轨道,形成四电子三中心d-pπ键,就是反馈配键,非金属配位化合物中也可能存在这种键。配位键可用以下三种理论来解释:
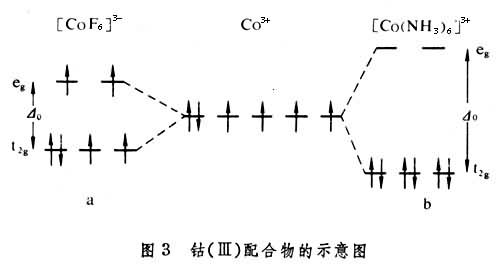
价键理论是由L.C.鲍林发展起来的。价键理论认为配体上的电子进入中心原子的杂化轨道。例如,钴(Ⅲ)的配合物可用图 1表示。
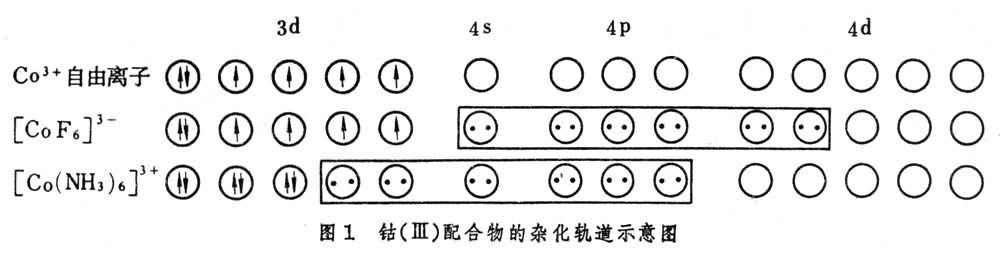
[CoF6]3-中F-的孤对电子进入Co3+的sp3d2杂化轨道,这种配合物称为外轨配合物或高自旋配合物。它与自由Co3+离子一样,有四个未成对电子,因而是顺磁性的。[Co(NH3)6]3+中NH3的孤对电子进入Co3+的d2sp3杂化轨道,这种配合物称为内轨配合物或低自旋配合物,由于所有电子都已成对,因而没有顺磁性而为抗磁性。
这个理论解释了配合物的立体化学和磁性质,但没有考虑激发态,也不能说明配合物的光谱等性质(见外轨配合物和内轨配合物)。
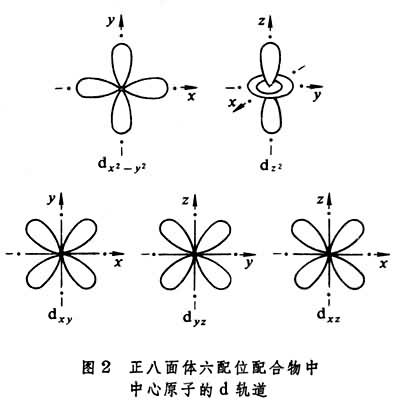
晶体场理论将配体看作点电荷或偶极子,来考虑配体产生的静电场对中心原子的原子轨道能级的影响。例如,把中心原子引入位于正八面体六个顶角上的六个配体中,原来五重简并的d轨道就分裂成一组二重简并的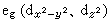 轨道和一组三重简并的t2g(dxy、dxz、dЖ)轨道。eg轨道直接指向位于顶角上的配体, 由于排斥作用,比不指向配体的t2g轨道有较高的能量(图2)。上述钴(Ⅲ)配合物可用图3表示。eg和t2g轨道的能量差,称为分离能墹0,墹0呏10Dq,Dq称为场强参量。6个F-产生的场不强,墹0较小,d电子按照洪德规则排布如图3a,有四个未成对电子,因而[CoF6]3-为弱场配合物或高自旋配合物。6个NH3产生的场较强,墹0较大,d电子按照能量最低原则和泡利原理排布如图3b,没有未成对电子,因而[Co(NH3)6]3+为强场配合物或低自旋配合物。理论上场强参量可用公式
轨道和一组三重简并的t2g(dxy、dxz、dЖ)轨道。eg轨道直接指向位于顶角上的配体, 由于排斥作用,比不指向配体的t2g轨道有较高的能量(图2)。上述钴(Ⅲ)配合物可用图3表示。eg和t2g轨道的能量差,称为分离能墹0,墹0呏10Dq,Dq称为场强参量。6个F-产生的场不强,墹0较小,d电子按照洪德规则排布如图3a,有四个未成对电子,因而[CoF6]3-为弱场配合物或高自旋配合物。6个NH3产生的场较强,墹0较大,d电子按照能量最低原则和泡利原理排布如图3b,没有未成对电子,因而[Co(NH3)6]3+为强场配合物或低自旋配合物。理论上场强参量可用公式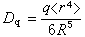 或
或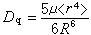 来计算,式中q和μ为配体的电荷和偶极矩;R 为中心原子和配体的矩离;〈r4〉为中心原子d轨道中的电子和原子核之间的距离的四次方的期望值。实际上,Dq常作为一种经验参量,由拟合光谱数据得出。
来计算,式中q和μ为配体的电荷和偶极矩;R 为中心原子和配体的矩离;〈r4〉为中心原子d轨道中的电子和原子核之间的距离的四次方的期望值。实际上,Dq常作为一种经验参量,由拟合光谱数据得出。
晶体场理论较合理地解释了配合物的磁性质和电子光谱,是研究配位化学最重要的理论。由于只考虑中心原子的电子结构,没有考虑配体的结构,不适用于烯烃配合物和零价金属配合物。
分子轨道理论假定电子是在分子轨道中运动,应用群论或根据成键的基本原则(对称性原则,能量相近原则和最大重叠原则)就可得出分子轨道能级图。再把电子从能量最低的分子轨道开始按照泡利原理逐一填入,即得分子的电子组态。分子轨道可以近似地用原子轨道线性组合而成。分子轨道可以分为成键轨道和反键轨道。分子的键合程度取决于分子中成键电子数与反键电子数之差。分子轨道理论更全面地讨论了配合物的结构,但计算工作量很大,常不得不引进各种不同程度的近似。