[拼音]:benbingtongsuanniaozheng
[外文]:phenylketonuria,PKU
苯丙氨酸羟化酶基因缺陷引起的遗传性氨基酸代谢障碍性疾病。常染色体隐性遗传。最早报告于1934年。临床特征是严重的智力发育障碍(白痴)及尿中有过量苯丙酮酸排出。对多数病例出生后单纯控制饮食中苯丙氨酸的摄入,即可成功地防止脑损害。新生儿患者可无症状,但若延误治疗一年,即可发生不可逆的脑损害,故早期诊断和治疗极为重要。发病率在世界各地差异很大,黑种人中罕见,在中国发病率为1:16000。
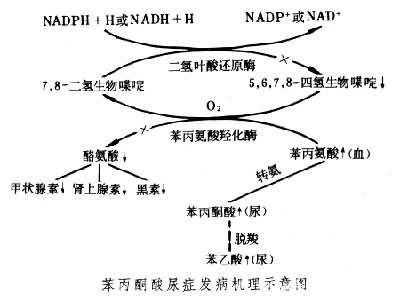
发病机理苯丙氨酸是必需氨基酸之一。人体摄入的苯丙氨酸除用于蛋白质合成外,在肝脏苯丙氨酸羟化酶及其辅因子的作用下生成酪氨酸,少量苯丙氨酸进入次要代谢途径,经转氨基作用变成苯丙酮酸。酪氨酸也是人体必需的氨基酸,可用以合成多巴胺、肾上腺素、甲状腺素等重要激素和神经递质及黑素,也可进一步代谢生成尿黑酸进入三羧酸循环为机体提供能量。有些人因遗传基因缺陷,其苯丙氨酸羟化酶(或辅因子)缺乏或活性减低,苯丙氨酸主要代谢途径受阻,血液中游离苯丙氨酸浓度异常升高,次要代谢途径活跃,致正常代谢时数量很少的产物苯丙酮酸在体液中明显增多,于是出现高苯丙氨酸血症及苯丙酮酸尿症(见图)。据信引起新生儿中枢神经系统分化停滞或发育迟缓的有害物质可能就是苯丙氨酸本身。发病机理是升高的苯丙氨酸与其他氨基酸在进入神经细胞时竞争转运系统,致神经细胞内氨基酸不平衡,从而抑制蛋白质合成及神经突触的形成,影响微管(纺锤丝的基本成分)的形成,造成大脑的不可逆损害。
苯丙氨酸可通过代谢旁路生成苯乳酸→苯乙酸→苯乙酰谷氨酰胺,这些代谢产物经汗及尿排出,使汗、尿带鼠尿或马粪气味。
分型及临床表现苯丙酮酸尿症患者的血液中苯丙氨酸增高。高苯丙氨酸血症分5型。经典PKU是Ⅰ型,此型病人出生时很少有症状,生后第1年的早期患儿身上可有类似鼠尿的霉臭味。苯丙氨酸的正常代谢产物酪氨酸减少,黑素合成缺乏必要的原料,故患儿毛发、虹膜及皮肤色素很淡。以上症状多不为双亲注意。1年以后出现明显的智力障碍、痴呆、不会走路及说话、多动、癫痫样发作、易激惹或有攻击行为。此外,还可出现多汗、流涎、震颤、小头畸型、牙釉质发育不良、生长速度减慢等。此型病人苯丙氨酸羟化酶几乎全无活性。Ⅱ、Ⅲ型病人尿中无苯丙酮酸,轻者无症状而重者似Ⅰ型。此二型的特点是苯丙氨酸食物耐量正常,并以此区别于典型的PKU。Ⅳ、Ⅴ型病例出生不久,即有吞咽困难,因而很难喂养,半年内生长明显迟钝、肌肉痉挛、多涎,1年内发生癫痫。其病因为苯丙氨酸羟化酶的天然辅因子再生系统有缺陷,二氢蝶啶还原酶缺乏,不仅苯丙氨酸羟化障碍,脑内5-羟色胺及左旋多巴形成也受严重影响,故神经系统合并症严重。
诊断出现典型症状后诊断不难,但脑损伤后作出诊断为时已晚,为及时作出诊断,唯一可靠的方法为在新生儿中筛选高苯丙氨酸血症病例。筛选成功的关键是:
(1)测定全部新生儿。
(2)在婴儿能摄入蛋白质(哺乳)后进行。因 PKU婴儿出生时血中苯丙氨酸可在正常水平。检查方法为格思里氏试验(血中含苯丙氨酸时,枯草杆菌的生长即不能为β-2-噻吩丙氨酸所抑制)。PKU与其他类型高苯丙氨酸血症的鉴别靠苯丙氨酸食物耐量。辅因子缺陷型病例的诊断需更特异的方法,包括酶学测定或补充相应辅因子后测血中苯丙氨酸水平。基因诊断技术的进步使PKU胎儿产前基因诊断成为可能。
治疗唯一切实可行的治疗方法是限制饮食中苯丙氨酸的摄入。市售低苯丙氨酸半合成食品可将每日苯丙氨酸摄入量限制在250~500mg之内。婴儿出生后60天内给予治疗者,体力和智力发育可正常。疗程要足够长,现倾向于 8~10岁后可慢慢放松严格控制,但若病情反复,则应重新恢复治疗。有辅因子缺陷的病例治疗困难,补充四氢生物蝶啶很难实现,因为它不能有效地穿过细胞或脑组织,单纯低苯丙氨酸饮食不能控制症状。PKU的女性患者妊娠期应继续治疗,否则可致婴儿小头畸型及智力迟钝。因苯丙氨酸降解主要在肝脏进行,故有人建议用肝移植的方法为患者提供稳定的酶源来治疗本病,这在将来也许可行。